Mit Hilfe der Immunhistologie lassen sich verschiedene Immunzellen (z.B. CD3, CD4, CD8, CD45RO, LFA1, Perforin pos. zytotoxische Zellen, Makrophagen – M1 und M2) in RNAlater-fixiertem Gewebe differenzieren und quantifizieren. Hierbei muss hervorgehoben werden, dass die Spezifität und Sensitivität der Immunhistologie bei der Verwendung von RNAlater fixiertem Gewebe gegenüber Formalin-fixiertem Gewebe deutlich verbessert ist (1,2,3,4,5,6).
Darüber hinaus werden weitere immunologische Parameter (z.B. HLA-DR, ICAM, VCAM, PAI-1) und die Quantität und Qualität der Fibrose (Gesamtkollagen, Kollagen I und III) analysiert (7,8,9,10).
Exakte Charakterisierung und Quantifizierung der Immunzellen bei gleichzeitigem Ausschluss einer Virusinfektion stellt die Grundlage für eine kausale immunsuppressive Therapie. Hierbei muss verdeutlicht werden, dass nicht nur CD3-positive Zellen als diagnostischer Parameter herangezogen werden sollten. Entgegen der aktuellen Guidelines konnten wir zeigen, dass bei über 25% der Patienten mit normaler CD3-Zellzahl weitere Immunzellen (CD45R0, LFA-1, Perforin-positive T-Zellen) signifikant erhöht sind (11).
Serviceangebote Immunhistochemie
- Gewebefixierung (RNAlater) und Lagerung nach GLP/GCP
- Anfertigung von Gefrierschnitten am Kryomikrotom
- Antikörperfärbungen
- Fluoreszenz- und Lichtmikroskopie
- Digitale Vermessung und Quantifizierung gefärbter Zellen oder Gewebeschnitte
- Befundung der Präparate
- Digitalbilder repräsentativer Gewebeabschnitte
- Archivierung der Befunde, Gewebeblöcke und Präparate
Zusätzliche Bestimmungen/Färbungen
- Färbung verschiedener Immunzellen (CD4+, CD8+)
- ARVC/D-Nachweis (Plakoglobin, Connexin 43, N-Cadherin)
- Spezialfärbungen bei Verdacht auf Speichererkrankungen: Amyloidose (Kongorot-Färbung), HCM, M.Fabry (CD77, Toluidinblau), Hämochomatose (Fe-Färbung)
- Endothelaktivierung (CD31, VCAM)
- Kollagenexpression (Col I-, Col III-, Sirius-Färbung)
- Makrophagen Differenzierung (M1, M2)
Nachweis der Amyloidose und Amyloid-Differenzierung / Subtypisierung
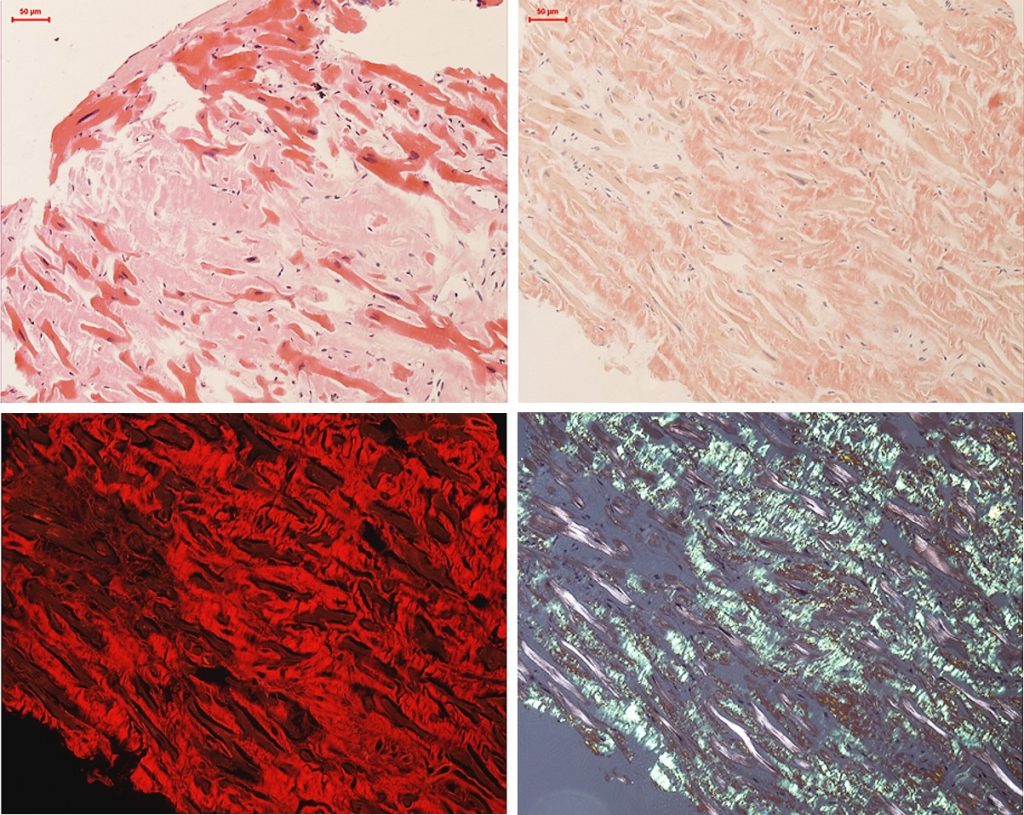
- Amyloidose ist eine seltene, aber verheerende Erkrankung, die durch die Ablagerung von fehlgefalteten Proteinen als Aggregate im extrazellulären Gewebe des Körpers verursacht wird und zu einer Beeinträchtigung der Organfunktion führt. Es wurde erkannt, dass ungefähr 25 Proteine Amyloidose verursachen, und dass das amyloidogene Protein die Grundlage für die derzeitige Klassifizierung ist. Es werden jedoch nur drei Amyloidoseformen im Herzmuskelgewebe gefunden: AA - Serum amyloid A, AL (Lambda and Kappa) und ATTR - Senile systemische (Herz-) Amyloidose (12).
Die Endomyokardbiopsie spielt eine zentrale Rolle für die Verifizierung bzw. Subtypisierung der kardialen Amyloidose. Deren frühzeitige Diagnose und kardial ausgerichtete vielversprechende Therapieoptionen sind aktuell von großer Bedeutung und stehen zunehmend im Fokus von Studien. Im Falle einer kardialen Amyloidose zeigt sich meist eine regelmäßige Verteilung der Amyloidablagerungen, sodass das Risiko für einen „sampling error“ und damit einem falsch negativen Resultat als relativ gering einzustufen ist. Amyloid wird durch Kongorotfärbung nachgewiesen, welches unter polarisiertem Licht betrachtet eine typische apfelgrüne / gelborange Doppelbrechung aufweist.
In der Biopsie sollte quantitativ die Amyloidlast bestimmt werden, da das bioptisch gesicherte Ausmaß der Amyloidose - vor allem bei der AL-Amyloidose - von entscheidender prognostischer und therapeutischer Bedeutung ist.
Es konnte gezeigt werden, dass der Nachweis einer intramyokardialen Entzündung für die Diagnose sowohl bei der Amyloidose als auch bei M.Farby von großer Bedeutung ist (13,14).
Darüber hinaus ist für die Therapieauswahl die exakte Klassifikation des Amyloids entscheidend. Diese kann durch immun-histochemische Färbungen mit hoher Sensitivität bereits in einem frühen Stadium der Erkrankung sicher erfolgen. Es gibt verlässliche Antikörper zum Nachweis der ATTR-, AL- und AA-Amyloidose, welche mehr als 99% der kardialen Amyloidosen ausmachen. Darüber hinaus konnten wir zeigen, dass eine zusätzliche Begleitentzündung prognostisch hoch relevant ist.
Im Falle des Nachweises einer ATTR-Amyloidose sollte eine genetische Untersuchung erfolgen, um eine sog. Altersamyloidose vom Wildtyp von der hereditären Form unterscheiden zu können.
Die immunhistochemische Typisierung des Amyloids erfolgt im IKDT.
Die Gendiagnostik der ATTR-Amyloidose erfolgt im UKSH (Institut für Pathologie, Prof. Dr. C.Röcken).
Nachweis der Arrhythmogenen rechtsventrikulären Dysplasie (ARVC)
- Genetisch bedingte arrhythmogene Herzmuskel-erkrankungen, wie die arrhythmogene rechtsventrikuläre Dysplasie (ARVC), können mit Hilfe des immunhistologischen Nachweises des Expressionsprofils desmosomaler Proteine (Plakoglobin, Desmoplakin, Caderin, Connexin 43), die im Falle einer ARVC vermindert exprimiert werden bzw. fehlen, diagnostiziert werden.
Es gibt zunehmend Hinweise darauf, dass auch bei genetisch bedingten Kardiomyopathie vor allem bei der HCM aber auch ARVD und M.Fabry die Inflammation bezüglich der Progression und dem Auftreten des plötzlichen Herztodes eine wichtige Rolle spielt.
Diagnostik bei Transplantationspatienten
Die Bewertung der Transplantatbstoßung erfolgt nach den internationalen Richtlinien der ISLHT mit Beurteilung der zellulären und Antikörper-vermittelten Abstoßungsreaktion. Zusätzlich führt das IKDT eine Charakterisierung der Mikrovaskulopathie zusammen mit einer immunhistochemische Entzündungsdiagnostik zum detaillierten Nachweis entzündlicher Infiltrate (Lymphozyten, Makrophagen, zytotoxischen Zellen, Adhäsionmoleküle) durch. Die einzige Technik, die eine detailgenaue Unterscheidung dieser verschiedenen Abstoßungsformen erlaubt, ist die Herzmuskelbiopsie. Sie ist auch heute noch der Goldstandard für die Feststellung von Abstoßungsreaktionen und wird entsprechend den Vorgaben der Fachgesellschaften eingesetzt.
Zelluläre Abstoßung
Die zelluläre Abstoßung ist durch das vermehrte Auftreten von Entzündungszellen (Lymphozyten, weiße Blutzellen) im Herzmuskelgewebe gekennzeichnet. Je nach Schweregrad der Abstoßung treten diese Entzündungszellen ungeordnet oder als „Haufen“ auf und sind mit bestimmten Gewebereaktionen verbunden. Dazu gehören eine Schwellung der Herzmuskelzellen, ntramyokardiale Blutungen oder auch Zelluntergänge.
Humorale Abstoßung
Die antikörper-vermittelte Abstoßung ist dadurch gekennzeichnet, dass sich körpereigene Botenstoffe (z.B. Komplementfaktoren) in Form von Eiweißen in den kleinen und kleinsten Gefäßen des Herzmuskelgewebes ablagern. Diese lösen dort eine Abwehrreaktion des Körpers aus.
Mikrovaskulopathie/Stenose
Eine weitere Möglichkeit zur Charakterisierung eines vorliegenden Abstoßungsprozesses ist die Beurteilung einer Vaskulopathie der kleinen Gefäße. Hierbei wird in einer gesonderten Bewertung das Verhältnis zwischen Lumenradius und der Wandstärke der Gefäße sowie das Verhältnis vom Durchmesser der Endothelzellen zur Dicke des Endotheliums analysiert.
Intramyokardiale Entzündung und Quilty-Phänomen
- Auch wenn die Richtlinien getreue Klassifikation keinen oder nur eine geringen Grad (ISHLT 0-1) der Abstoßung ergibt, findet sich oft parallel eine teils massive Infiltration von Entzündungszellen, die eine Ursache für eine klinische Verschlechterung des Patienten sein könnte. Ein diesbezüglicher Befund ermöglicht der Transplantationsklinik eine kurzzeitige therapeutische Intervention zur Reduktion der Entzündungszellen, zusätzlich zur Standardmedikation für Transplantierte. Neben dem oben beschriebenen Entzündungsgeschehen gibt es eine Ansammlung von Entzündungszellen, ausschließlich in der innersten Schicht des Herzmuskels, das Quilty-Phänomen. Während dies früher als Nebeneffekt der Immunsuppression angenommen wurde, wird dieser Befund heute als eine mögliche Vorstufe der zellulären Abstoßung diskutiert (15,16).
- Imanaka-Yoshida K. Inflammation in myocardial disease: From myocarditis to dilated cardiomyopathy. Pathol Int. 2020 Jan;70(1):1-11. doi: 10.1111/pin.12868. Epub 2019 Nov 5. PMID: 31691489. https://pubmed.ncbi.nlm.nih.gov/31691489/
- Bracamonte-Baran W, Čiháková D. Cardiac Autoimmunity: Myocarditis. Adv Exp Med Biol. 2017;1003:187-221. doi: 10.1007/978-3-319-57613-8_10. PMID: 28667560; PMCID: PMC5706653. https://pubmed.ncbi.nlm.nih.gov/28667560/
- Ohta-Ogo K, Sugano Y, Ogata S, Nakayama T, Komori T, Eguchi K, Dohi K, Yokokawa T, Kanamori H, Nishimura S, Nakamura K, Ikeda Y, Nishimura K, Takemura G, Anzai T, Hiroe M, Hatakeyama K, Ishibashi-Ueda H, Imanaka-Yoshida K. Myocardial T-Lymphocytes as a Prognostic Risk-Stratifying Marker of Dilated Cardiomyopathy - Results of the Multicenter Registry to Investigate Inflammatory Cell Infiltration in Dilated Cardiomyopathy in Tissues of Endomyocardial Biopsy (INDICATE Study). Circ J. 2022 Jun 24;86(7):1092-1101. doi: 10.1253/circj.CJ-21-0529. Epub 2022 Mar 10. PMID: 35264513. https://pubmed.ncbi.nlm.nih.gov/35264513/
- Escher F, Kühl U, Lassner D, Stroux A, Westermann D, Skurk C, Tschöpe C, Poller W, Schultheiss HP. Presence of perforin in endomyocardial biopsies of patients with inflammatory cardiomyopathy predicts poor outcome. Eur J Heart Fail. 2014 Oct;16(10):1066-72. doi: 10.1002/ejhf.148. Epub 2014 Aug 28. PMID: 25163698. https://pubmed.ncbi.nlm.nih.gov/25163698/
- Escher F, Kühl U, Lassner D, Stroux A, Gross U, Westermann D, Pieske B, Poller W, Schultheiss HP. High Perforin-Positive Cardiac Cell Infiltration and Male Sex Predict Adverse Long-Term Mortality in Patients With Inflammatory Cardiomyopathy. J Am Heart Assoc. 2017 Aug 18;6(8):e005352. doi: 10.1161/JAHA.116.005352. PMID: 28862949; PMCID: PMC5586411. https://pubmed.ncbi.nlm.nih.gov/28862949/
- Baumeier C, Escher F, Aleshcheva G, Pietsch H, Schultheiss HP. Plasminogen activator inhibitor-1 reduces cardiac fibrosis and promotes M2 macrophage polarization in inflammatory cardiomyopathy. Basic Res Cardiol. 2021 Jan 11;116(1):1. doi: 10.1007/s00395-020-00840-w. PMID: 33432417; PMCID: PMC7801308. https://pubmed.ncbi.nlm.nih.gov/33432417/
- Noutsias M, Seeberg B, Schultheiss HP, Kühl U. Expression of cell adhesion molecules in dilated cardiomyopathy: evidence for endothelial activation in inflammatory cardiomyopathy. Circulation. 1999 Apr 27;99(16):2124-31. doi: 10.1161/01.cir.99.16.2124. PMID: 10217652. https://pubmed.ncbi.nlm.nih.gov/10217652/
- Vallbracht KB, Schwimmbeck PL, Seeberg B, Kühl U, Schultheiss HP. Endothelial dysfunction of peripheral arteries in patients with immunohistologically confirmed myocardial inflammation correlates with endothelial expression of human leukocyte antigens and adhesion molecules in myocardial biopsies. J Am Coll Cardiol. 2002 Aug 7;40(3):515-20. doi: 10.1016/s0735-1097(02)01990-3. PMID: 12142120. https://pubmed.ncbi.nlm.nih.gov/12142120/
- Westermann D, Lindner D, Kasner M, Zietsch C, Savvatis K, Escher F, von Schlippenbach J, Skurk C, Steendijk P, Riad A, Poller W, Schultheiss HP, Tschöpe C. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Fail. 2011 Jan;4(1):44-52. doi: 10.1161/CIRCHEARTFAILURE.109.931451. Epub 2010 Nov 12. PMID: 21075869. https://pubmed.ncbi.nlm.nih.gov/21075869/
- Baumeier C, Escher F, Aleshcheva G, Pietsch H, Schultheiss HP. Plasminogen activator inhibitor-1 reduces cardiac fibrosis and promotes M2 macrophage polarization in inflammatory cardiomyopathy. Basic Res Cardiol. 2021 Jan 11;116(1):1. doi: 10.1007/s00395-020-00840-w. PMID: 33432417; PMCID: PMC7801308. https://pubmed.ncbi.nlm.nih.gov/33432417/
- Baumeier C, Harms D, Aleshcheva G, Gross U, Escher F, Schultheiss HP. Advancing Precision Medicine in Myocarditis: Current Status and Future Perspectives in Endomyocardial Biopsy-Based Diagnostics and Therapeutic Approaches. J Clin Med. 2023 Jul 31;12(15):5050. doi: 10.3390/jcm12155050. PMID: 37568452; PMCID: PMC10419903. https://pubmed.ncbi.nlm.nih.gov/37568452/
- Garcia-Pavia P, Rapezzi C, Adler Y, Arad M, Basso C, Brucato A, Burazor I, Caforio ALP, Damy T, Eriksson U, Fontana M, Gillmore JD, Gonzalez-Lopez E, Grogan M, Heymans S, Imazio M, Kindermann I, Kristen AV, Maurer MS, Merlini G, Pantazis A, Pankuweit S, Rigopoulos AG, Linhart A. Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2021 Apr 21;42(16):1554-1568. doi: 10.1093/eurheartj/ehab072. PMID: 33825853; PMCID: PMC8060056. https://pubmed.ncbi.nlm.nih.gov/33825853/
- Siegismund CS, Escher F, Lassner D, Kühl U, Gross U, Fruhwald F, Wenzel P, Münzel T, Frey N, Linke RP, Schultheiss HP. Intramyocardial inflammation predicts adverse outcome in patients with cardiac AL amyloidosis. Eur J Heart Fail. 2018 Apr;20(4):751-757. doi: 10.1002/ejhf.1039. Epub 2017 Oct 25. PMID: 29067795. https://pubmed.ncbi.nlm.nih.gov/29067795/
- Frustaci A, Verardo R, Grande C, Galea N, Piselli P, Carbone I, Alfarano M, Russo MA, Chimenti C. Immune-Mediated Myocarditis in Fabry Disease Cardiomyopathy. J Am Heart Assoc. 2018 Sep 4;7(17):e009052. doi: 10.1161/JAHA.118.009052. PMID: 30371172; PMCID: PMC6201436. https://pubmed.ncbi.nlm.nih.gov/30371172/
- Ludhwani D, Abraham J, Kanmanthareddy A. Heart Transplantation Rejection. 2022 Sep 19. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan–. PMID: 30725742. https://pubmed.ncbi.nlm.nih.gov/30725742/
- Coutance G, Desiré E, Duong Van Huyen JP. A Review of Biomarkers of Cardiac Allograft Rejection: Toward an Integrated Diagnosis of Rejection. Biomolecules. 2022 Aug 18;12(8):1135. doi: 10.3390/biom12081135. PMID: 36009029; PMCID: PMC9405997. https://pubmed.ncbi.nlm.nih.gov/36009029/